Parkinson's disease
| Parkinson's | |
|---|---|
| Classification and external resources | |
 Illustration of the Parkinson disease by Sir William Richard Gowers from A Manual of Diseases of the Nervous System in 1886 |
|
| ICD-10 | G20., F02.3 |
| ICD-9 | 332 |
| DiseasesDB | 9651 |
| MedlinePlus | 000755 |
| eMedicine | neuro/304 neuro/635 in young pmr/99 rehab |
Parkinson's disease (also known as Parkinson's, Parkinson disease or PD) is a degenerative disorder of the central nervous system that often impairs the sufferer's motor skills, speech, and other functions.[1]
Parkinson's disease belongs to a group of conditions called movement disorders. It is characterized by muscle rigidity, tremor, postural abnormalities, gait abnormalities, a slowing of physical movement (bradykinesia) and a loss of physical movement (akinesia) in extreme cases. The primary symptoms are the results of decreased stimulation of the motor cortex by the basal ganglia, normally caused by the insufficient formation and action of dopamine produced in the dopaminergic neurons of the midbrain (specifically the substantia nigra). Secondary symptoms may include high level cognitive dysfunction and subtle language problems. PD is both chronic and progressive.
PD is the most common cause of chronic progressive parkinsonism, a term which refers to the syndrome of tremor, rigidity, bradykinesia and postural instability. PD is also called "primary parkinsonism" or "idiopathic PD" (classically meaning having no known cause). While many forms of parkinsonism are idiopathic, "secondary" cases may result from toxicity most notably of drugs, head trauma, or other medical disorders. The disease is named after English apothecary James Parkinson, who made a detailed description of the disease in his essay: "An Essay on the Shaking Palsy" (1817).
Contents |
Classification
The term Parkinsonism is used for a motor syndrome whose main symptoms are tremor at rest, stiffness, slowing of movement and postural instability. Parkinsonisms can be divided into four subtypes according to their origin: primary or idiopathic (no known origin), secondary or acquired, hereditary parkinsonism, and parkinson plus syndromes or multiple system degeneration.[1]
"Parkinson's disease" is the most common form of parkinsonism, and refers to the normal presentation of idiopathic parkinsonisms.[1][2] Genetic forms are usually included although the terms familial Parkinson’s disease and familial parkinsonism are also used for disease entities with an autosomal dominant or recessive pattern of inheritance.[3] Parkinson-plus diseases are primary parkinsonisms which present additional features.[3] They include multiple system atrophy (MSA), progressive supranuclear palsy (PSP), corticobasal degeneration (CBD) and dementia with Lewy bodies (DLB).[3] While idiopathic Parkinson's disease patients also have Lewy bodies in their brain tissue, the distribution is denser and more widespread in DLB. Even so, the relationship between Parkinson disease, Parkinson disease with dementia (PDD), and dementia with Lewy bodies (DLB) might be most accurately conceptualized as a spectrum, with a discrete area of overlap between each of the three disorders.
Signs and symptoms
Parkinson's disease affects movement, producing motor symptoms.[1] Non-motor symptoms, which include autonomic dysfunction, cognitive and neurobehavioral problems, and sensory and sleep difficulties, are also common but are under-appreciated.[1]
Motor
Four motor symptoms are considered cardinal in PD: tremor, rigidity, bradykinesia and postural instability.[1] Tremor is the most apparent and well-known symptom.[1] It is most commonly a rest tremor: maximal when the limb is at rest and disappearing with voluntary movement and sleep.[1] It affects to a greater extent the most distal part of the extremity and is typically unilateral at onset becoming bilateral later.[1] Though around 30% of PD sufferers do not have tremor at disease onset most of them would develop it along the course of the disease.[1] Rigidity is due to joint stiffness and increased muscle tone, which combined with a resting tremor produce a ratchety, "cogwheel rigidity" when the limb is passively moved.[1] Rigidity may be associated with joint pain, such pain being a frequent initial manifestation of the disease.[1] Bradykinesia (slowness of movement) is the most characteristic clinical feature of PD and it produces difficulties not only with the execution of a movement but also with its planning and initiation.[1] The performance of sequential and simultaneous movements is also hindered.[1] Bradykinesia is the most disabling symptom in the beginning of the disease.[3] In the late stages of the disease postural instability is typical, which leads to impaired balance and falls.[1] Nevertheless instability many times is absent in the initial stages, specially in younger patients.[3]
PD motor symptomatology is not limited to these four symptoms. Gait and posture disturbances such as decreased arm swing, a forward-flexed posture and the use of small steps when walking; speech and swallowing disturbances; and other symptoms such as a mask-like face expression or a small handwriting are only examples of the ample range of common motor problems that can appear with the disease.[1]
Neuropsychiatric
Parkinson's disease causes neuropsychiatric disturbances, which include mainly cognition, mood and behavior problems and can be as disabling as motor symptoms.[1]
Cognitive disturbances occur even in the initial stages of the disease in some cases.[4] A very high proportion of sufferers will have mild cognitive impairment as the disease advances.[1] Most common cognitive deficits in non-demented patients are executive dysfunction, which translates into impaired set shifting, poor problem solving, and fluctuations in attention among other difficulties; slowed cognitive speed, memory problems; specifically in recalling learned information, with an important improvement with cues; and visuospatial skills difficulties, which are seen when the person with PD is for example asked to perform tests of facial recognition and perception of line orientation.[4]
Deficits tend to aggravate with time, developing in many cases into dementia. A person with PD has a sixfold increased risk of suffering dementia,[1] and the overall rate in people with the disease is around 30%.[4] Moreover, prevalence of dementia increases in relation to disease duration, going up to 80%.[4] Dementia has been associated with a reduced quality of life in disease sufferers and caregivers, increased mortality and a higher probability of attending a nursing home.[4]
Cognitive problems and dementia are usually accompanied by behavior and mood alterations, although these kind of changes are also more common in those patients without cognitive impairment than in the general population. Most frequent mood difficulties include depression, apathy and anxiety.[1] Obsessive–compulsive behaviors such as craving, binge eating, hypersexuality, pathological gambling, or other, can also appear in PD, and have been related to a dopamine dysregulation syndrome associated with the medications for the disease.[1] Psychotic symptoms are common in later PD. Symptoms of psychosis are either hallucinations, or delusions.[5]
Other
In addition to cognitive and motor symptoms PD can impair other body functions. Sleep problems can be worsened by medications for PD, but they are a core feature of the disease.[1] They can manifest as excessive daytime somnolence, disturbances in REM sleep or insomnia.[1] The autonomic system is altered which can lead for example to orthostatic hypotension, oily skin and seborrheic dermatitis, excessive sweating, urinary incontinence and altered sexual function.[1] Constipation and gastric dysmotility can be severe enough to endanger comfort and health.[6] PD is also related to different ophthalmological abnormalities such as decreased blink rate and alteration in the tear film, leading to irritation of the eye surface, abnormalities in ocular pursuit and saccadic movements and limitations in the upward gaze.[1] Changes in perception include reduced sense of smell and sensation of pain and paresthesias.[1]
Causes
Most people with Parkinson's disease are described as having idiopathic Parkinson's disease (having no specific known cause). A small proportion of cases is known to be originated in genetic factors.
Genetic
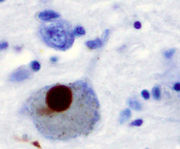
PD traditionally has been considered a non-genetic disorder, however around 15% of patients have a first-degree relative who also has the disease.[3] At least between 5 and 10% of the patients are now known to have monogenic forms of the disease.[7] Other genes act as risk factors for sporadic cases of the disease.
A number of specific genetic mutations causing Parkinson's disease have been discovered. Genes conclusively identified are Alpha-synuclein (SNCA), ubiquitin carboxy-terminal hydrolase L1 (UCH-L1), parkin (PRKN), leucine-rich repeat kinase 2 (LRRK2 or dardarin) , PTEN-induced putative kinase 1 (PINK1), DJ-1 and ATP13A2.[7][8] With the exception of LRRK2 they account for a small minority of cases of PD.[8] Most studied PD-related genes are SNCA and LRRK2.
The role of the SNCA gene is central in the pathophysiology of PD, since the Alpha-synuclein protein is the main component of Lewy bodies, which are present in this and other diseases.[7] Missense mutations of the gene and duplications and triplications of the locus containing it have been found in different groups with familial PD.[7] Missense mutations are very rare with few families found in all the world, most with a common ancestor.[7] On the other hand multiplications of the SNCA locus account for around 2% of familial cases.[7] Additionally multiplications have age-dependent or incomplete penetrance, since they have also been found in non symptomatic carriers.[7]
The LRRK2 gene (PARK8) encodes for a protein called dardarin. The name dardarin comes from a basque word for tremor since such gene was first identified in families from England and the north of Spain.[8] LRRK is the most common cause known of familial and sporadic PD, with mutations in this gene in up to 10% of the patients with a family history of the disease and 3% of sporadic cases.[7][8] Although more than 40 different mutations of the gene have been found, one of them (G2019S) appears in 5% of the familial cases and 2% of the sporadic ones in Europe.[7] Nevertheless prevalence of this mutation varies greatly with ethnicity, being for example rare in Asia but very common in PD cases of northern Africa and among Ashkenazi Jews[7] The penetrance of the G2019S mutation ranges between 28% at age 60 and 75% at age 80 with sex having no effect.[7]
Other mutations in at least three of the causative genes of the famililial forms of the disease have been found to be a risk factor for the so-called sporadic PD. The three are SNCA, LRRK2 and GBA.[7] Genome-wide association studies; which search for mutated alleles with low penetrance in sporadic cases; have yielded few positive results although the number of such studies has been scarce and their size small.[7]
Pathophysiology



The main pathological characteristic is cell death in the substantia nigra and more specifically the ventral part of the pars compacta, affecting up to 70% of the cells by death time.[8] The mechanisms by which the brain cells in Parkinson's are lost are varied.[9] One mechanism consists of an abnormal accumulation of the protein alpha-synuclein bound to ubiquitin in the damaged cells. This protein accumulation forms inclusions called Lewy bodies.[8] According to the Braak staging, a classification of the disease based on pathological findings, Lewy bodies first appear in the olfactory bulb, medulla oblongata and pontine tegmentum, being at this stage patients asymptomatic. As the disease evolves Lewy bodies later attain the substantia nigra, areas of the midbrain and basal prosencephalon and finally reach areas of the neocortex.[8] Other cell-death mechanisms include proteosomal and lysosomal system dysfunction, and reduced mitochondrial activity.[9]
There are four major dopamine pathways in the brain; the nigrostriatal pathway, referred to above, mediates movement and is the most conspicuously affected in early Parkinson's disease. The other pathways are the mesocortical, the mesolimbic, and the tuberoinfundibular. Disruption of dopamine along the non-striatal pathways likely explains much of the neuropsychiatric pathology associated with Parkinson's disease.
The direct pathway facilitates movement and the indirect pathway inhibits movement, thus the loss of these cells leads to a hypokinetic movement disorder. The lack of dopamine results in increased inhibition of the ventral anterior nucleus of the thalamus, which sends excitatory projections to the motor cortex, thus leading to hypokinesia The symptoms of Parkinson's disease result from the greatly reduced activity of pigmented dopamine-secreting (dopaminergic) cells in the pars compacta region of the substantia nigra (literally "black substance"). These neurons project to the striatum and their loss leads to alterations in the activity of the neural circuits within the basal ganglia that regulate movement. In essence, GABA/ Substance P of the direct pathways diminish, leading to less inhibition of the pars reticulata and internal globus palidus and an inhibition of the indirect pathway by way of GABA/ enkaphalins.
Excessive accumulations of iron, which are toxic to nerve cells, are also typically observed in conjunction with the protein inclusions. Iron and other transition metals such as copper bind to neuromelanin in the affected neurons of the substantia nigra. Neuromelanin may be acting as a protective agent. The most likely mechanism is generation of reactive oxygen species.[10][11] Iron also induces aggregation of synuclein by oxidative mechanisms.[12] Similarly, dopamine and the byproducts of dopamine production enhance alpha-synuclein aggregation. The precise mechanism whereby such aggregates of alpha-synuclein damage the cells is not known. The aggregates may be merely a normal reaction by the cells as part of their effort to correct a different, as-yet unknown, insult. Based on this mechanistic hypothesis, a transgenic mouse model of Parkinson's has been generated by introduction of human wild-type alpha-synuclein into the mouse genome under control of the platelet-derived-growth factor-β promoter.[13]
A recent view of Parkinson's disease implicates specialized calcium channels that allow substantia nigra neurons, but not most neurons, to repetitively fire in a "pacemaker" like pattern. The consequent flooding of calcium into these neurons may aggravate damage to mitochondria and may cause cell death. One study has found that, in experimental animals, treatment with a calcium channel blocker isradapine had a substantial protective effect against the development of Parkinson's disease.[14]
Diagnosis

Typically, the diagnosis is based on medical history and neurological examination.[1] The physician interviews and observes the patient in the search of the cardinal motor symptoms of the disease, while also attending to other symptoms that would exclude the diagnosis of PD.[1] Response to levodopa is another sign pointing towards PD.[1] However there is no definitive test for diagnosis although finding lewy bodies during autopsy has been traditionally considered the gold standard for diagnosis.[1] Common presentations of the disease are usually easily diagnosed, however the disease can be difficult to diagnose accurately, especially in its early stages, since parkinsonisms can occur due to a range of pathologies differing from PD.[1]
Medical organizations have created diagnostic criteria to ease and standardize the diagnostic process especially in the first stages of the disease. The most widely known are those by the UK Parkinson’s Disease Society Brain Bank and the National Institute of Neurological Disorders and Stroke.[1] For example the PD Society Brain Bank criteria require in the first place the person to suffer from slowness of movement (bradykinesia) and either rigidity, rest tremor or postural instability. Secondly other possible causes for symptoms have to be ruled out. Finally the person would have to have 3 or more of the following during onset or evolution: unilateral onset, rest tremor, progression, asymmetry of motor symptoms, response to levodopa during at least 5 years, clinical course of at least ten years and appearance of dyskinesias induced by the intake of excessive levodopa.[1] Accuracy of diagnostic criteria evaluated at autopsy is between 75 and 90% with specialists such as neurologists having higher rates.[1]
Differential diagnosis includes other kind of tremors and also other causes of parkinsonism.[15] Other tremors include postural and action tremors or intention tremor.[15] Other causes that secondarily produce a parkinsonian syndrome are Alzheimer's disease, multiple cerebral infarction, and drug induced parkinsonism.[15] Parkinson plus syndromes such as progressive supranuclear palsy and multiple system atrophy also have to be ruled out.[1] Anti-Parkinson's medications are typically less effective controlling symptoms in Parkinson-plus diseases.[1] Faster progression rates, early cognitive dysfunction or postural instability, minimal tremor or exccesive symmetry at onset may also indicate a Parkinson plus disease.[2]
Computed tomography (CT) and Magnetic resonance imaging (MRI) brain scans of people with PD usually appear normal.[16] These techniques are nevertheless useful for diagnosis to rule out other diseases which can be secondary causes of parkinsonism such as basal ganglia tumors, vascular pathology and hydrocephalus.[16] A specific technique of MRI, diffusion MRI, has been reported to be useful at discriminating between typical and atypical parkinsonism, although its exact diagnostic value is still under investigation.[16] Dopaminergic function in the basal ganglia can be measured with the help of different PET and SPECT radiotracers. Examples are ioflupane (123I) (trade name DaTSCAN) and iometopane (Dopascan) for SPECT or 18F for PET.[16] A pattern of reduced dopaminergic activity in the basal ganglia, can aid in diagnosing PD.[16]
Management
At present, there is no cure for Parkinson's disease, but medications, surgery and multidisciplinary management can provide relief from the symptoms.
PD is a chronic disorder that requires broad-based management including patient and family education, support group services, general wellness maintenance, physiotherapy, exercise, and nutrition.
Main families of drugs useful for motor symptoms of PD are Levodopa, dopamine agonists and MAO-B inhibitors.[17] Regarding medications the treatment approach varies depending on the stage of the person with the disease. Two phases are usually considered: an initial phase in which the PD sufferer has already developed some disability for which he needs pharmacological treatment, and a second stage in which the patient develops motor complications related to levodopa usage.[17] Treatment in the initial state aims to attain an equilibrium between reduced dopaminergic therapy and good management of symptoms. Additionally L-DOPA beginning is delayed using other medications such as MAO-B inhibitors and dopamine agonists so the later PD stage is retarded.[17] In the second stage the aim is to reduce symptoms while controlling fluctuations between on and off periods. Sudden withdrawals from medication, and overuse by some patients, also have to be controlled.[17]
Levodopa

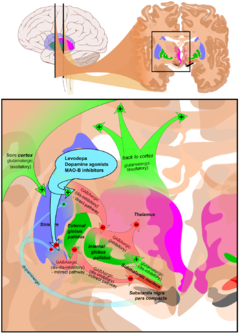
Levodopa (or L-DOPA) has been the most widely used treatment for over 30 years.[17] L-DOPA is transformed into dopamine in the dopaminergic neurons by dopa-decarboxylase.[17] Since motor symptoms of PD are produced by a lack of dopamine in the substantia nigra the administration of L-DOPA temporarilly diminishes the motor symptomatology.[17]
Only 5-10% of L-DOPA crosses the blood-brain barrier. The remaining L-DOPA is often metabolised to dopamine elsewhere, causing a wide variety of side effects including nausea, dyskinesias and stiffness.[17] Carbidopa and benserazide are peripheral dopa decarboxylase inhibitors.[17] They help to prevent the metabolism of L-DOPA before it reaches the dopaminergic neurons and therefore reduce side effects. They are generally given as combination preparations with levodopa.[17] Existing preparations are carbidopa/levodopa (co-careldopa, trade names Sinemet, Parcopa, Atamet) and benserazide/levodopa (co-beneldopa, trade name Madopar).
There are controlled release versions of Sinemet and Madopar that spread out the effect of the levodopa. Duodopa is a combination of levodopa and carbidopa. Slow-release levodopa preparations have not shown an increased control of motor symptoms or motor complications when compared to immediate release preparations.[17]
Tolcapone inhibits the COMT enzyme, which degrades dopamine, thereby prolonging the effects of levodopa.[17] It has been used to complement levodopa. However, due to its possible side effects such as liver failure, it's limited in its availability.[17] A similar drug, entacapone has not been shown to cause significant alterations of liver function and maintains adequate inhibition of COMT over time.[17] Entacapone is available for treatment alone (COMTan) or combined with carbidopa and levodopa (Stalevo).[17]
Due to feedback inhibition, levodopa results in a reduction in the endogenous formation of L-DOPA, and so eventually becomes counterproductive. Levodopa preparations lead in the long term to the development of motor complications characterized by involuntary movements called dyskinesias and fluctuations in the response to medication.[17] When this occurs PD sufferers change fastly from stages with good response to medication and few symptoms ("on" state) to phases with no response to medication and important motor symptoms ("off" state).[17] For this reason levodopa doses are maintained as low as possible while maintaining functionality.[17] Delaying the initiation of dopatherapy, using instead alternatives for some time, is also common practice.[17] A former strategy to reduce motor complications was to withdraw patients from L-DOPA for some time. It is discouraged now since it can bring dangerous side effects such as neuroleptic malignant syndrome.[17] Most people will eventually need levodopa and later develop motor complications.[17]
Dopamine agonists
Dopamine agonists in the brain have a similar effect to levodopa since they bind to dopaminergic post-synaptic receptors.[17] Dopamine agonists were initially used for patients experiencing on-off fluctuations and dyskinesias as a complementary therapy to levodopa but they are now mainly used on their own as an initial therapy for motor symptoms with the aim of delaying motor complications.[17][18] When used in late PD they are useful at reducing the off periods.[17] Dopamine agonists include bromocriptine, pergolide, pramipexole, ropinirole , piribedil, cabergoline, apomorphine, and lisuride.
Agonists produce significant, although mild, side effects including somnolence, hallucinations, insomnia, nausea, and constipation.[17] Sometimes side effects appear even at a the minimal clinically efficacious dose, leading the physician to search for a different agonist or kind of drug.[17] When compared with levodopa, while they delay motor complications they control worse symptoms.[17] Nevertheless they are usually effective enough to manage symptoms in the initial years of the disease.[3] They are also more expensive than levodopa.[3] Dyskinesias with dopamine agonists are rare in younger patients, but along other side effects more common in older patients.[3] All this has led to agonists being the preferential initial treatment for the former as opposed to levodopa in the latter.[3]
Apomorphine, which is a non-orally administered dopamine agonist, may be used to reduce off periods and dyskinesia in late PD.[17] It is administered by intermitent injections or continuous subcutaneous infusions.[17] Since secondary effects such as confusion and hallucinations are not rare it has been recommended that patients under apomorphine treatment should be closely monitored.[17]
MAO-B inhibitors
MAO-B inhibitors (Selegiline and rasagiline) increase the level of dopamine in the basal ganglia by blocking its metabolization. They inhibit monoamine oxidase-B (MAO-B) which breaks down dopamine secreted by the dopaminergic neurons. Therefore reducing MAO-B results in higher quantities of L-DOPA in the striatum.[17] Similarly to dopamine agonists, MAO-B inhibitors improve motor symptoms and delay the need of taking levodopa when used as monotherapy in the first stages of the disease but produce more adverse effects and are less effective than levodopa. Evidence on their efficacy in the advanced stage is reduced although it points towards them being useful to reduce fluctuations between on and off periods.[17] Although an initial study had as result that selegiline in combination with levodopa increased the risk of death this has been later disproven.[17]
Other drugs
There is some indication that other drugs such as amantadine and anticholinergics may be useful as treatment of motor symptoms in early and late PD, but since quality of evidence on efficacy is reduced they are not first choice treatments.[17] In addition to motor PD is accompanied by an ample range of different symptoms. Different compounds are used to improve some of these problems.[19] Examples are the use of clozapine for psychosis, cholinesterase inhibitors for dementia and modafinil for day somnolence.[19][20]
Surgery and deep brain stimulation

Treating motor symptomatology in Parkinson's disease with surgery was once a common practice, but after the discovery of levodopa, number of surgeries was reduced.[21] Studies in the past few decades have led to great improvements in surgical techniques, and surgery is again being used in people with advanced PD for whom drug therapy is no longer sufficient.[21] Deep brain stimulation (DBS) is presently the most used surgical mean of treatment but other surgical therapies consisting in producing lesions in specific subcortical areas are also effective.[21] DBS involves the implantation of a medical device called a brain pacemaker, which sends electrical impulses to specific parts of the brain. Target areas for DBS or lesions include the thalamus, the globus pallidus (the lesion technique being called pallidotomy) or the subthalamic nucleus.[21]
Rehabilitation
There is partial evidence that speech or mobility problems can improve with rehabilitation although studies are still scarce and of low quality.[22][23] Regular physical exercise and/or therapy can be beneficial to the patient for maintaining and improving mobility, flexibility, strength, gait speed, and quality of life;[23] Exercise may also improve constipation.[6] One of the most widely practiced treatment for speech disorders associated with Parkinson's disease is the Lee Silverman Voice Treatment (LSVT), which focuses on increasing vocal loudness and has an intensive approach of one month.[22][24] Speech therapy and specifically LSVT may improve voice and speech function.[22] Occupational therapy (OT) aims to promote health and quality of life by helping people with the disease to participate in as many activities of their daily living as possible.[22] Number of studies and quality of them regarding the effectiveness of OT is poor, although there is some indication that it may improve motor skills and quality of life for the duration of the therapy.[22][25]
Diet
Muscles and nerves that control the digestive process may be affected by PD, therefore, it is common for patients to experience constipation and gastroparesis (food remaining in the stomach for a longer period of time than normal).[6] A balanced diet is hence recommended to help improve digestion. Diet should include high-fiber foods and plenty of water.[6] Levodopa and proteins use the same transportation system in the intestine and the blood-brain barrier, competing between them for access.[6] When taken together the consequences of such competition is a reduced effectiveness of the drug.[6] Therefore when levodopa is introduced excessive proteins are discouraged, while in advanced stages additional intake of low-protein products such as bread or pasta is recommended for similar reasons.[6] To minimize interaction with proteins levodopa is recommended to be taken 30 minutes before meals.[6] At the same time, regimens for PD patients restrict proteins during breakfast and lunch and are usually taken at dinner.[6] As the disease advances dysphagia may appear. In such cases specific measures include the use of thickening agents for liquid intake, special postures when eating and gastrostomy in the worst cases.[6]
Alternative treatments
Different nutrients have been proposed as possible treatments of PD; however there is no evidence that vitamin or food additives improve PD symptoms.[26] There is not enough evidence to suggest that acupuncture, and practice of qigong or tai chi have any effect on PD symptoms.[27][28][29] Fava and velvet beans are natural sources of L-DOPA and are taken by many patients. While they have shown some effectiveness,[30] their intake is not free of risks. Life threatening adverse reactions have been described, such as the neuroleptic malignant syndrome.[31][32]
Prognosis

PD progresses with time. If not treated motor symptoms have an aggressive advance at the beginning of the disease with an slower advance later on the disease course: untreated patients are expected to lose independent ambulation after 8 years and be bedridden after 10 years.[33] However it is not common to find untreated people nowadays and medication has improved the prognosis of motor symptoms, while at the same time it is also a new source of disability due to the undesired effects of L-DOPA after years of use.[33] Different studies of the history of the disease on people taking L-DOPA have found that the mean progression of symptoms to a stage of high dependency of subjects may take around 15 years.[33] However it is hard to predict what course the disease will take for an individual.[33] Age is the best predictor of disease progression.[9] Rate of motor decline is greater in those with less impairment at diagnosis while cognitive impairment is more frequent in those who are over 70 years of age at symptoms onset.[9]
Since current therapies improve motor symptoms, disability at present is mainly related to non-motor features of the disease.[9] Nevertheless the relationship between disease progression and disability is not linear. At first disability is related to motor symptoms and specially motor complications, which appear in up to 50% of the patients after 5 years of L-DOPA usage.[33] As the disease advances disability is more related to motor symptoms that have a bad response to medication such as swallowing and speech difficulties and gait and balance problems.[33] Finally after ten years most people with the disease suffer autonomic disturbances, sleep problems, mood alterations and cognitive decline.[33] All of them, but specially the latter, greatly increase disability.[33][9]
The average life expectancy of a PD patient is lower than for people who do not have the disease.[33] Mortality ratios are around twice of those unaffected.[33] Cognitive decline and dementia, old age at onset, a more advanced disease, and presence of swallowing problems are all mortality risk factors. Conversely a cause of death twice as common in PD patients than in the healthy population is aspiration pneumonia.[33] On the other hand a disease mainly characterized by tremor as opposed to rigidity predicts an improved survival.[33]
Epidemiology

Two main measures are used in epidemiological studies: incidence and prevalence. Incidence is the number of new cases per unit of person–time at risk (usually number of new cases per thousand person–years); while prevalence is the total number of cases of the disease in the population at a given time. PD is the most common neurodegenerative disorder after Alzheimer's disease.[34] The prevalence is estimated at being 0.3 in the whole population in industrialized countries, rising to 1% in those over 60 years of age and to 4% of the population over 80.[34] Mean age of onset is around 60 years, although 5-10% of cases are considered of young onset as begin between the age of 20 and 50.[3] Parkinson's disease might less prevalent in those of African and Asian ancestry, although results are controversial.[34] Some studies have also proposed that PD is more common in men than women while others did not found any differences between the two groups.[34] Studies on incidence report that it is between 8 and 18 per 100.000 person-years.[34]
Many risk and protective factors have been proposed in accordance to possible mechanisms of the disease, however none have been conclusively related to PD. While many different epidemiological studies have been carried out trying to prove or disprove the relationship between a given factor and PD, they have commonly been biased, and results of different studies have been contradictory.[34] Most replicated relationships are an increased risk of PD in those living in rural environments and those exposed to pesticides; and a reduced risk in smokers.[34]
Risk and protective factors
MPTP injections produce a range of symptoms similar to PD as well as a selective damage to the dopaminergic neurons in the substantia nigra. This has led to theorizing that exposure to some environmental toxins may increase the risk of suffering PD.[34] Toxins that have been consistently related to the disease are certain pesticides and herbicides, with exposure doubling the risk of having PD.[34] Conversely indirect measures of exposure to such toxins, such as living in rural environments have also been found to increasing the risk of PD. Heavy metals exposure has also been proposed to increase the risk of PD, being its hypothetical mechanism their accumulation in the substantia nigra during the course of the disease, however studies on the issue have been inconclusive.[34]
A protective effect of tobacco has been consistently found in epidemiological studies. The basis for such effect is not known but possibilities include an effect of nicotine as dopamine stimulant.[34] Caffeine consumption has also been proposed to protect from PD, but the hypothesis requires further validation.[34] Antioxidants, such as vitamin C and D, have been proposed to protect against the disease but results of studies have been contradictory and their positive effect is not proven. Regarding fat and fatty acids, the results on their relationship with PD have been contradictory, with both protective, risk enhancing or no effects.[34] Finally there have also been some preliminary indications of a possible protective role of estrogens and anti-inflammatory drugs.[34]
History

The disease was not formally recognized and its symptoms were not documented until 1817 in An Essay on the Shaking Palsy[35] by the British apothecary James Parkinson. Parkinson's disease was then known as paralysis agitans (shaking palsy in English), the term "Parkinson's disease" being coined later by Jean-Martin Charcot.[36]
Early descriptions
There are some early sources which talk about symptoms that are coherent with PD.[37] An Egyptian papyrus from the 12th century B.C talks about a king drooling with age and the Bible has references to tremor.[36][37] An early ayurvedic medical treatise dating back to the 10th century B.C describes a disease that evolves with tremor, lack of movement, drooling and other symptoms of PD. Moreover, this disease was treated with remedies derived from the mucuna family, which is rich in L-DOPA.[37] Galen writes on a disease which most surely is PD when he describes tremors that occur only at rest, postural changes and paralysis.[37]
After Galen there are no references known to be related to PD until the 17th century.[37] In this and the following century different authors write about some of the elements of the disease, preceding the description by Parkinson. Franciscus Sylvius separated as Galen tremor at rest from other tremors, and Johannes Baptiste Sagar and Hieronymus David Gaubius talk about festination.[37][38] John Hunter provided an excellent description of the disease, and it is even thought that gave Parkinson the idea of collecting and describing patients with "paralisis agitans".[37][39] Finally, Auguste François Chomel in his pathology treatise, which is contemporary to Parkinson's essay, has many descriptions on abnormal movements and rigidity typical of PD.[37]
XIXth century

In 1817 James Parkinson, aged 62, publishes his essay on the disease, reporting 6 cases of paralysis agitans.[36] In "An Essay of the Shaking Palsy" he describes the patients' resting tremor, abnormal posture and gait, paralysis and diminished muscle strength.[40] The accuracy of the description of the disease course is also remarkable.[36] He also acknowledges the contributions of many of the previous authors to the understanding of PD.[36] Although the article has been later considered the first seminal work on the disease, it received very little attention for more than forty years.[40] Nevertheless early neurologists who made further additions to the knowledge of the disease include Trousseau, Charcot, Gowers, Kinnier Wilson and Erb.[36] Moreover Charcot studies between 1868 and 1881 were a landmark in the understanding of the disease.[36] Among other advances he made the distinction between rigidity, weakness and bradykinesia.[36] He also led the disease to be re-named in behalf of James Parkinson.[36]
XXth century
First speculations of the anatomical substrate of PD were made 80 years after Parkinson's essay, when Brissaud proposed that the disease had its origin in the subthalamus or the cerebral peduncle and might be caused by an ischemic lesion.[36] In 1912 Frederic Lewy described a pathologic finding in affected brains, later named "Lewy bodies".[36] It was Konstantin Tretiakoff who in 1919 discovered that the substantia nigra was the main cerebral structure affected, but it was not widely accepted until further studies by Hassler in 1938.[36] The underlying biochemical changes in the brain were identified in the 1950s due largely to the work of Arvid Carlsson on the neurotransmitter dopamine and his role on PD. He later won a Nobel Prize for his work.[41]
While levodopa was first synthesized in 1911 by Funk, it received little attention until the mid 20th century.[41] It entered clinical practice in 1967, and the first large study reporting improvements in patients with Parkinson's disease resulting from treatment with levodopa was published in 1968. Levodopa use was a major revolution in the management of PD which still lasts.[41][42] Surgery for tremor was initiated in 1939 and was improved for over 20 years, but the arrival of levodopa reduced its use dramatically.[43] By the late 1980s deep brain stimulation emerged as a possible treatment and it was approved by the FDA in 1997.[44]
Research directions
Animal models
The tragedy of a group of drug addicts in California in the early 1980s who consumed a contaminated and illicitly produced batch of the synthetic opiate MPPP brought to light MPTP as a cause of PD symptoms.[45] Other predominant toxin-based models employ the insecticide rotenone, the herbicide paraquat and the fungicide maneb.[46] Models based on toxins are most commonly used in primates. Transgenic rodent models also exist.[47]
Gene therapy
Gene therapy is currently under investigation.[9][48] It involves the use of a non-infectious virus to shuttle a gene into a part of the brain. The gene used leads to the production of an enzyme which helps to manage PD symptoms or protects the brain from further damage.[9] As of 2010 there are four clinical trials using gene therapy in PD.[9] There have not been important adverse effects in these trials although the clinical usefulness of gene therapy is still unknown.[9]
Neuroprotective treatments
Investigations on neuroprotection are at the forefront of PD research. Several molecules have been proposed as potential treatments.[9] However none of them has been conclusively demonstrated to reduce degeneration in clinical trials.[9] Agents currently under investigation as neuroprotective agents include anti-apoptotic drugs (TCH346, CEP-1347), antiglutamatergic agents, monoamine oxidase inhibitors (selegiline, rasagiline), promitochondrial drugs (coenzyme Q10, creatine), calcium channel blockers (isradipine) and growth factors (GDNF).[9]
Neural transplantation
Cell transplants in PD started around 1980 using very different tissues such as fetal, porcine, carotid or retinal.[9] Although there was initial evidence of mesencephalic dopamine-producing cell transplants being beneficial, the best constructed studies up to date indicate that cell transplants have no effect.[9] An additional significant problem was the excess release of dopamine by the transplanted tissue, leading to dystonias.[49] Stem cells transplants have raised great recent interest. When transplanted into the brains of rodents and monkeys they survive and improve behavioral abnormalities.[9][50] Nevertheless while fetal stem cells are the easiest to manipulate their use is controversial.[9] Such controversy may be overcome with the use of induced pluripotent stem cells from adults.[9]
Society and culture

In 1957, William Black, President of Chock full o'Nuts coffee company, founded the Parkinson's Disease Foundation (PDF) after one of his company's employees was diagnosed with Parkinson's. Black launched the organization with a $250,000 grant to support Parkinson's Research.[51] While at first a regional organization, PDF expanded the scope of its activities throughout the U.S., and merged with the United Parkinson Foundation in 1999. Today, PDF focuses on funding research to learn the causes of and find a cure for Parkinson's, collaborating on initiatives to increase participation in Parkinson's disease clinical research such as PDtrials, as well as providing education and support for people with Parkinson's in the U.S. Since it was founded in 1957, PDF has provided more than $85 million for research, as well as $30 million for education and advocacy programs.[52]
The American Parkinson Disease Association was founded in 1961.[53] The European Parkinson's Disease Association, founded in 1992, is an organization based in Europe that supports Parkinson's disease research.
Actor Michael J. Fox, whose book, Lucky Man (2000), focused on his experiences with the disease and his career and family travails in the midst of it, established The Michael J. Fox Foundation for Parkinson's Research to develop a cure for Parkinson's disease. Another foundation that supports Parkinson's research was established by professional cyclist Davis Phinney. The Davis Phinney Foundation strives to improve the lives of those living with Parkinson's disease.
April 11, birthday of James Parkinson, is the world's Parkinson's disease day.[36]
References
- ↑ 1.00 1.01 1.02 1.03 1.04 1.05 1.06 1.07 1.08 1.09 1.10 1.11 1.12 1.13 1.14 1.15 1.16 1.17 1.18 1.19 1.20 1.21 1.22 1.23 1.24 1.25 1.26 1.27 1.28 1.29 1.30 1.31 1.32 1.33 1.34 1.35 Jankovic J (April 2008). "Parkinson's disease: clinical features and diagnosis". J. Neurol. Neurosurg. Psychiatr. 79 (4): 368–76. doi:10.1136/jnnp.2007.131045. PMID 18344392. http://jnnp.bmj.com/content/79/4/368.full.
- ↑ 2.0 2.1 Poewe W, Wenning G (November 2002). "The differential diagnosis of Parkinson's disease". Eur. J. Neurol. 9 Suppl 3: 23–30. PMID 12464118.
- ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 Samii A, Nutt JG, Ransom BR (May 2004). "Parkinson's disease". Lancet 363 (9423): 1783–93. doi:10.1016/S0140-6736(04)16305-8. PMID 15172778.
- ↑ 4.0 4.1 4.2 4.3 4.4 Caballol N, Martí MJ, Tolosa E (September 2007). "Cognitive dysfunction and dementia in Parkinson disease". Mov. Disord. 22 Suppl 17: S358–66. doi:10.1002/mds.21677. PMID 18175397.
- ↑ Friedman JH (June 2010). "Parkinson's disease psychosis 2010: A review article". Parkinsonism Relat Disord. doi:10.1016/j.parkreldis.2010.05.004. PMID 20538500.
- ↑ 6.0 6.1 6.2 6.3 6.4 6.5 6.6 6.7 6.8 6.9 Barichella M, Cereda E, Pezzoli G (October 2009). "Major nutritional issues in the management of Parkinson's disease". Mov. Disord. 24 (13): 1881–92. doi:10.1002/mds.22705. PMID 19691125.
- ↑ 7.00 7.01 7.02 7.03 7.04 7.05 7.06 7.07 7.08 7.09 7.10 7.11 7.12 Lesage S, Brice A (April 2009). "Parkinson's disease: from monogenic forms to genetic susceptibility factors". Hum. Mol. Genet. 18 (R1): R48–59. doi:10.1093/hmg/ddp012. PMID 19297401.
- ↑ 8.0 8.1 8.2 8.3 8.4 8.5 8.6 Davie CA (2008). "A review of Parkinson's disease". Br. Med. Bull. 86: 109–27. doi:10.1093/bmb/ldn013. PMID 18398010.
- ↑ 9.00 9.01 9.02 9.03 9.04 9.05 9.06 9.07 9.08 9.09 9.10 9.11 9.12 9.13 9.14 9.15 9.16 9.17 Obeso JA, Rodriguez-Oroz MC, Goetz CG, et al. (May 2010). "Missing pieces in the Parkinson's disease puzzle". Nat Med 16 (6): 653–61. doi:10.1038/nm.2165. PMID 20495568.
- ↑ Jenner P (1998). "Oxidative mechanisms in nigral cell death in Parkinson's disease". Movement Disorders 13 (Suppl 1): 24–34. PMID 9613715.
- ↑ Chiueh CC, Andoh T, Lai AR, Lai E, Krishna G (2000). "Neuroprotective strategies in Parkinson's disease: protection against progressive nigral damage induced by free radicals". Neurotoxicity Research 2 (2-3): 293–310. doi:10.1007/BF03033799. PMID 16787846.
- ↑ Kaur D, Andersen JK (October 2002). "Ironing out Parkinson's disease: is therapeutic treatment with iron chelators a real possibility?". Aging Cell 1 (1): 17–21. doi:10.1046/j.1474-9728.2002.00001.x. PMID 12882349.
- ↑ Masliah E, Rockenstein E, Veinbergs I, et al (2000). "Dopaminergic loss and inclusion body formation in alpha-synuclein mice: implications for neurodegenerative disorders". Science 287 (5456): 1265–9. doi:10.1126/science.287.5456.1265. PMID 10678833.
- ↑ Chan CS, Guzman JN, Ilijic E, et al. (June 2007). "'Rejuvenation' protects neurons in mouse models of Parkinson's disease". Nature 447 (7148): 1081–6. doi:10.1038/nature05865. PMID 17558391.
- ↑ 15.0 15.1 15.2 The National Collaborating Centre for Chronic Conditions, ed (2006). "Diagnosing Parkinson's Disease". Parkinson's Disease. London: Royal College of Physicians. pp. 29–47. ISBN 1-86016-283-5.
- ↑ 16.0 16.1 16.2 16.3 16.4 Brooks DJ (April 2010). "Imaging approaches to Parkinson disease". J. Nucl. Med. 51 (4): 596–609. doi:10.2967/jnumed.108.059998. PMID 20351351.
- ↑ 17.00 17.01 17.02 17.03 17.04 17.05 17.06 17.07 17.08 17.09 17.10 17.11 17.12 17.13 17.14 17.15 17.16 17.17 17.18 17.19 17.20 17.21 17.22 17.23 17.24 17.25 17.26 17.27 17.28 17.29 17.30 17.31 17.32 17.33 The National Collaborating Centre for Chronic Conditions, ed (2006). "Symptomatic pharmacological therapy in Parkinson’s disease". Parkinson's Disease. London: Royal College of Physicians. pp. 59–100. ISBN 1-86016-283-5.
- ↑ Goldenberg MM (October 2008). "Medical management of Parkinson's disease". P & T 33 (10): 590–606. PMID 19750042.
- ↑ 19.0 19.1 The National Collaborating Centre for Chronic Conditions, ed (2006). "Non-motor features of Parkinson’s disease". Parkinson's Disease. London: Royal College of Physicians. pp. 113–133. ISBN 1-86016-283-5.
- ↑ Hasnain M, Vieweg WV, Baron MS, Beatty-Brooks M, Fernandez A, Pandurangi AK (July 2009). "Pharmacological management of psychosis in elderly patients with parkinsonism". Am. J. Med. 122 (7): 614–22. doi:10.1016/j.amjmed.2009.01.025. PMID 19559160.
- ↑ 21.0 21.1 21.2 21.3 The National Collaborating Centre for Chronic Conditions, ed (2006). "Surgery for Parkinson’s disease". Parkinson's Disease. London: Royal College of Physicians. pp. 101–111. ISBN 1-86016-283-5.
- ↑ 22.0 22.1 22.2 22.3 22.4 The National Collaborating Centre for Chronic Conditions, ed (2006). "Other key interventions". Parkinson's Disease. London: Royal College of Physicians. pp. 135–146. ISBN 1-86016-283-5.
- ↑ 23.0 23.1 Goodwin VA, Richards SH, Taylor RS, Taylor AH, Campbell JL (April 2008). "The effectiveness of exercise interventions for people with Parkinson's disease: a systematic review and meta-analysis". Movement Disorders 23 (5): 631–40. doi:10.1002/mds.21922. PMID 18181210.
- ↑ Fox CM, Ramig LO, Ciucci MR, Sapir S, McFarland DH, Farley BG (November 2006). "The science and practice of LSVT/LOUD: neural plasticity-principled approach to treating individuals with Parkinson disease and other neurological disorders". Seminars in Speech and Language 27 (4): 283–99. doi:10.1055/s-2006-955118. PMID 17117354.
- ↑ Dixon L, Duncan D, Johnson P, et al. (2007). "Occupational therapy for patients with Parkinson's disease". Cochrane Database Syst Rev (3): CD002813. doi:10.1002/14651858.CD002813.pub2. PMID 17636709.
- ↑ Suchowersky O, Gronseth G, Perlmutter J, Reich S, Zesiewicz T, Weiner WJ (April 2006). "Practice Parameter: neuroprotective strategies and alternative therapies for Parkinson disease (an evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology". Neurology 66 (7): 976–82. doi:10.1212/01.wnl.0000206363.57955.1b. PMID 16606908.
- ↑ Lee MS, Lam P, Ernst E (December 2008). "Effectiveness of tai chi for Parkinson's disease: a critical review". Parkinsonism Relat. Disord. 14 (8): 589–94. doi:10.1016/j.parkreldis.2008.02.003. PMID 18374620.
- ↑ Lee MS, Ernst E (January 2009). "Qigong for movement disorders: A systematic review". Mov. Disord. 24 (2): 301–3. doi:10.1002/mds.22275. PMID 18973253.
- ↑ Lee MS, Shin BC, Kong JC, Ernst E (August 2008). "Effectiveness of acupuncture for Parkinson's disease: a systematic review". Mov. Disord. 23 (11): 1505–15. doi:10.1002/mds.21993. PMID 18618661.
- ↑ Katzenschlager R, Evans A, Manson A, et al (2004). "Mucuna pruriens in Parkinson's disease: a double blind clinical and pharmacological study". J. Neurol. Neurosurg. Psychiatr. 75 (12): 1672–7. doi:10.1136/jnnp.2003.028761. PMID 15548480.
- ↑ Ladha SS, Walker R, Shill HA (May 2005). "Case of neuroleptic malignant-like syndrome precipitated by abrupt fava bean discontinuance". Mov. Disord. 20 (5): 630–1. doi:10.1002/mds.20380. PMID 15719433.
- ↑ Raguthu L, Varanese S, Flancbaum L, Tayler E, Di Rocco A (October 2009). "Fava beans and Parkinson's disease: useful 'natural supplement' or useless risk?". Eur. J. Neurol. 16 (10): e171. doi:10.1111/j.1468-1331.2009.02766.x. PMID 19678834.
- ↑ 33.00 33.01 33.02 33.03 33.04 33.05 33.06 33.07 33.08 33.09 33.10 33.11 Poewe W (December 2006). "The natural history of Parkinson's disease". J. Neurol. 253 Suppl 7: VII2–6. doi:10.1007/s00415-006-7002-7. PMID 17131223.
- ↑ 34.00 34.01 34.02 34.03 34.04 34.05 34.06 34.07 34.08 34.09 34.10 34.11 34.12 34.13 de Lau LM, Breteler MM (June 2006). "Epidemiology of Parkinson's disease". Lancet Neurol 5 (6): 525–35. doi:10.1016/S1474-4422(06)70471-9. PMID 16713924.
- ↑ Parkinson J (2002). "An essay on the shaking palsy. 1817". The Journal of Neuropsychiatry and Clinical Neurosciences 14 (2): 223–36; discussion 222. doi:10.1176/appi.neuropsych.14.2.223. PMID 11983801.
- ↑ 36.00 36.01 36.02 36.03 36.04 36.05 36.06 36.07 36.08 36.09 36.10 36.11 36.12 Lees AJ (September 2007). "Unresolved issues relating to the shaking palsy on the celebration of James Parkinson's 250th birthday". Mov. Disord. 22 Suppl 17: S327–34. doi:10.1002/mds.21684. PMID 18175393.
- ↑ 37.0 37.1 37.2 37.3 37.4 37.5 37.6 37.7 García Ruiz PJ (December 2004). "Prehistoria de la enfermedad de Parkinson" (in Spanish; Castilian). Neurologia 19 (10): 735–7. PMID 15568171. http://www.arsxxi.com/Revistas/fframesart.php?MTk%3D&NTUzNw%3D%3D&MA%3D%3D&U1A%3D&QVJUSUNVTE8gREUgUEFHTw%3D%3D&MTk%3D&NDcx.
- ↑ Koehler PJ, Keyser A (September 1997). "Tremor in Latin texts of Dutch physicians: 16th-18th centuries". Mov. Disord. 12 (5): 798–806. doi:10.1002/mds.870120531. PMID 9380070.
- ↑ Currier RD (April 1996). "Did John Hunter give James Parkinson an idea?". Arch. Neurol. 53 (4): 377–8. PMID 8929162.
- ↑ 40.0 40.1 Louis ED (November 1997). "The shaking palsy, the first forty-five years: a journey through the British literature". Mov. Disord. 12 (6): 1068–72. doi:10.1002/mds.870120638. PMID 9399240.
- ↑ 41.0 41.1 41.2 Fahn S (2008). "The history of dopamine and levodopa in the treatment of Parkinson's disease". Mov. Disord. 23 Suppl 3: S497–508. doi:10.1002/mds.22028. PMID 18781671.
- ↑ Hornykiewicz O (2002). "L-DOPA: from a biologically inactive amino acid to a successful therapeutic agent". Amino Acids 23 (1-3): 65–70. doi:10.1007/s00726-001-0111-9. PMID 12373520.
- ↑ Guridi J, Lozano AM (November 1997). "A brief history of pallidotomy". Neurosurgery 41 (5): 1169–80; discussion 1180–3. doi:10.1097/00006123-199711000-00029. PMID 9361073.
- ↑ Coffey RJ (March 2009). "Deep brain stimulation devices: a brief technical history and review". Artif Organs 33 (3): 208–20. doi:10.1111/j.1525-1594.2008.00620.x. PMID 18684199.
- ↑ Langston JW, Ballard P, Tetrud JW, Irwin I (February 1983). "Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis". Science 219 (4587): 979–80. doi:10.1126/science.6823561. PMID 6823561.
- ↑ Cicchetti F, Drouin-Ouellet J, Gross RE (September 2009). "Environmental toxins and Parkinson's disease: what have we learned from pesticide-induced animal models?". Trends Pharmacol. Sci. 30 (9): 475–83. doi:10.1016/j.tips.2009.06.005. PMID 19729209.
- ↑ Harvey BK, Wang Y, Hoffer BJ (2008). "Transgenic rodent models of Parkinson's disease". Acta Neurochir. Suppl. 101: 89–92. doi:10.1007/978-3-211-78205-7_15. PMID 18642640.
- ↑ Feng, LR, Maguire-Zeiss KA (2010). "Gene Therapy in Parkinson's Disease: Rationale and Current Status". CNS Drugs 24 (3): 177–92. doi:10.2165/11533740-000000000-00000. PMID 20155994.
- ↑ Redmond DE (October 2002). "Cellular replacement therapy for Parkinson's disease--where we are today?". The Neuroscientist 8 (5): 457–88. doi:10.1177/107385802237703. PMID 12374430.
- ↑ ""Stem Cell Research Aims to Tackle Parkinson's Disease"". http://www.sciencedaily.com/releases/2006/12/061204123212.htm. Retrieved 2010-04-16.
- ↑ "Education: Joy in Giving". Time. 1960-01-18. http://www.time.com/time/magazine/article/0,9171,828597,00.html. Retrieved 2010-03-01.
- ↑ "About PDF". http://www.pdf.org/en/about_pdf. Retrieved 2010-08-09.
- ↑ http://www.apdaparkinson.org/
External links
- Parkinson's Disease at the Open Directory Project
- Parkinson's Disease: Hope Through Research (National Institute of Neurological Disorders and Stroke)
- World Parkinson Disease Association
- GeneReview/NIH/UW entry on LRRK2-Related Parkinson Disease
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
||||||||||||||||||||||||||||||||||